The
systematic study of coordination compounds was started by a very famous Swiss
scientist Alfred Werner whose pioneering work opened an entirely new
field of investigation in inorganic chemistry. He prepared and characterized a
large number of coordination compounds and studied their physical, chemical and
isomeric behaviour by simple experimental techniques. On the basis of these
studies. Werner, in 1898, propounded his theory of coordination compounds. Which
is later termed
as Werner’s Theory of Coordinate Compounds. Due to this theory he
is awarded by Nobel prize and he is also called the ‘Father
of Coordination Chemistry’.

Tuesday, August 20, 2019
Monday, August 12, 2019
CRYSTAL FIELD SPLITTING IN OCTAHEDRALCOMPLEXES:
For
convenience, let us assume that the six ligands are positioned symmetrically
along the Cartesian axes, with the metal atom at the origin. As the ligands
approach, first there is an increase in the energy of d orbitals to that of the
free ion just as would be the case in a spherical filed. Next, the orbitals
lying along the axes (dz2and dx2-y2 d)
get repelled more strongly than dxy, dyz and dxz
orbitals, which have lobes directed between the axes. The dxy , dyz , dxz
orbitals are lowered in energy relative to the average energy in the spherical
crystalfiled.
Thus, the degenerate set of d orbitals get split into two sets: the lower
energy orbitals set, t2g and the higher energy, eg set.
The energy separation is denoted by del.oct (the subscript o is for octahedral.

Crystal field stabilisation energy (CFSE):
The difference in energy of eg and t2g
Orbitals are called crystal field stabilisation energy
(CFSE):

Where
m
and n = are
number of electrons in t2g
and eg
orbitals respectively and del.oct
is crystalfield splitting energy in octahedral Complexes.
l =
represents the number of extra electron pair formed because of the ligands in
comparison to normal degenerate configuration.
P= (Pairing
energy) the energy required for electron pairing in a single orbital.
The actual configuration of complex adopted is decided by the relative values
of delta and P
Case (1):
If del.oct
is less than P
We have so called weak field or high spin situation, the fourth electron
entered one of the eg orbitals giving configuration (t2g3
and eg1) If now 5th
electron is added to a weak field the configuration become (t2g3
and eg2).
Case (2):
If del.oct
is more than P , we have the strong field , low spin situation and
pairing will occur in the t2g
level with eg
level remaining unoccupied in entities of d1 and d6 ions .
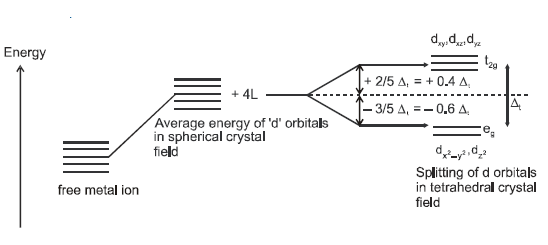
CRYSTAL FIELD SPLITTING IN TETRAHEDRAL COMPLEXES:
Tetahedral
complex (sp3):
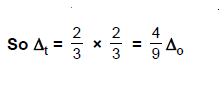
OTHER EXAMPLES :
In a
tetrahedral field : Consider a cube such that a metal atom or ion is situated
at its centre of symmetry through which the axis of geometry are passing and
joining the face centres of this cube. Therefore, lobes of eg orbitals will be directed
towards the face centres but those of t2g orbitals will be pointing towards
edge centres. Now consider 4 monodentate ligands approaching the metal, the 4 alternate
corners of this cube so as to make a tetrahedron.
Thus it is
clear that t2g
orbitals are nearer to the ligands than the eg orbitals. Hence t2g orbitals
will experience more repulsion than eg orbitals. Therefore, crystal field
splitting will be reversed of octahedral field which can be shown as below.
In
tetrahedral complexes none of the ligand is directly facing any orbital so the
splitting is found to be small in comparison to octahedral complexes. For the
same metal, the same ligands and metal-ligand distances, it can be shown that del.tetra = (4/9) del.oct.
This may attributes to the following two reasons.
(1) There are only four ligands instead of six, so
the ligand field is only two thirds the size; as the ligand field spliting is
also the two thirds the size and
(2) The
direction of the orbitals does not concide with the direction of the ligands.
This reduces the crystal field spliting by roughly further two third.
Consequently,
the orbital splitting energies are not sufficiently large for forcing pairing
and, therefore, low spin configurations are rarely observed.
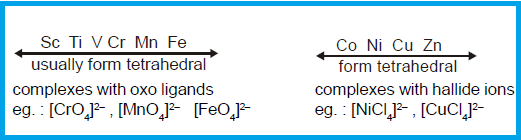
FACTORS FAVOURING TETRAHEDRAL COMPLEXES:
Tetrahedral
complexes are favoured by steric requirements, either simple electrostatic repulsion
of charge ligands or vander wall's repulsions of large one. A valence bond (VB)
point of view ascribed tetrahedral structure to sp3 hybridisation.
Tetrahedral
complexes are thus generally favoured by large ligands like Cl-, B-, I-
and PPh3 and metal ions of
six types;
(1) Those with a noble gas configuration
such as Be2+ (ns0);
(2) Those with pseudo noble gas
configuration (n-1)
d10ns0np0, such as Zn2+, Cu+ and Ga3+, and
(3) Those transition metal ions which do
not strongly favour other structure by virtue of the CFSE, such as Co2+, d7.
(4) Those transition metal which have
lower oxidation state.
(5) Those metals generally with
electronic configuration d0, d5 and d10 prefer
to form such complexes.
(6) It is observed that
|
SN
|
Complex
|
Nature
|
|
1
|
[Ni(CO)4]
|
Diamagnetic
|
|
2
|
[Ni(Cl)4]2-
|
Paramagnetic with two unpaired
electron
|
|
3
|
[NiCl2(pph3)2]
|
Paramagnetic with two unpaired
electron
|
|
4
|
[MnCl4]2-
|
Paramagnetic with five unpaired
electron
|
|
5
|
[FeCl4]2-
|
Paramagnetic with four unpaired
electron
|
|
6
|
[Cu(py)4]+
|
Diamagnetic
|
|
7
|
Cs2[CuCl4]
|
Paramagnetic with two unpaired
electron (Orange tetrahedral) Sp3
|
|
8
|
NH3[CuCl4]
|
Paramagnetic with two unpaired
electron (Yellow Square Planer) dsp2
|
|
9
|
[Zn(NH3)4]2+
|
(d10) CFSE=0 , Diamagnetic
|
|
10
|
[Zn(CN)4]2-
|
(d10) CFSE=0 , Diamagnetic
|
Subscribe to:
Posts (Atom)